Trimming and Filtering reads
Overview
Teaching: 30 min
Exercises: 0 minQuestions
What steps are required for basic data cleaning in RNA-seq studies?
Objectives
Understand the various data cleaning steps for RNA-seq data.
Learn how to perfrorm adapter removal and quality trimming.
Cleaning Reads
In the previous section, we took a high-level look at the quality of each of our samples using FastQC. We visualized per-base quality graphs showing the distribution of read quality at each base across all reads in a sample and extracted information about which samples fail which quality checks. Some of our samples failed quite a few quality metrics used by FastQC. This doesn’t mean, though, that our samples should be thrown out! It’s very common to have some quality metrics fail, and this may or may not be a problem for your downstream application.
Adapter removal
- “Adapters” are short DNA sequences that are added to each read as part of the sequencing process (we won’t get into “why” here).
- These are removed as part of the data generation steps that occur during the sequencing run, but sometimes there is still a non-trivial amount of adapter sequence present in the FASTQ files.
- Since the sequence is not part of the target genome (i.e., the genome if the species from which teh samples were derived) then we need to remove it to prevent it affecting the downstream analysis.
- The FastQC application get detection adapter contamination in samples.
We will use a program called CutAdapt to filter poor quality reads and trim poor quality bases from our samples.
How to act on fastq after QC.
We can do several trimming:
- on quality using Phred score. What will be the Phred score?
- on the sequences, if they contain adaptor sequences.
To do so, we can use on tools: The cutadapt application is often used to remove adapter sequence from FASTQ files.
- The following syntax will remove the adapter sequence AACCGGTT from the file SRR014335-chr1.fastq, create a new file called SRR014335-chr1_trimmed.fastq, and write a summary to the log file SRR014335-chr1.log:
$ pwd
/home/[Your_Username]/obss_2021/RNA_seq
$ mkdir Trimmed
$ module load cutadapt/2.10-gimkl-2020a-Python-3.8.2
$ cutadapt -q 20 -a AACCGGTT -o Trimmed/SRR014335-chr1_cutadapt.fastq Raw/SRR014335-chr1.fastq > Trimmed/SRR014335-chr1.log
We can have a look at the log file to see what cutadapt has done.
$ less Trimmed/SRR014335-chr1.log
Now we should trim all samples.
$ cd Raw
$ ls
SRR014335-chr1.fastq SRR014336-chr1.fastq SRR014337-chr1.fastq SRR014339-chr1.fastq SRR014340-chr1.fastq SRR014341-chr1.fastq
$ for filename in *.fastq
> do base=$(basename ${filename} .fastq)
> cutadapt -q 20 -a AACCGGTT -o ../Trimmed/${base}.trimmed.fastq ${filename} > ../Trimmed/${base}.log
> done
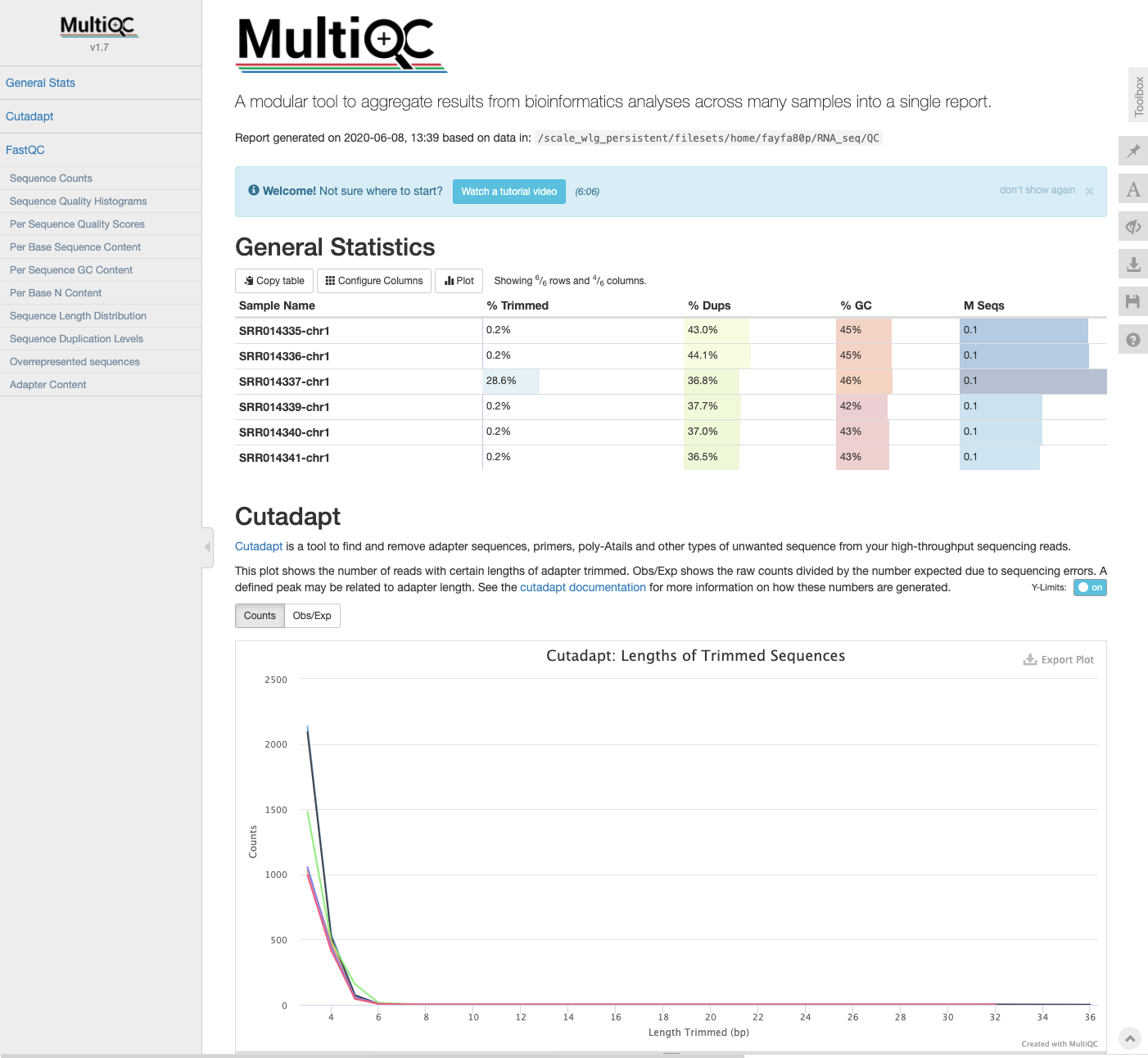
MultiQC: cutadapt log files
- If the log files from
cutadaptare added to the directory containing the FastQC output, this information will also be incorporated into the MultiQC report the next time it is run.
$ cd ../MultiQC
$ cp ../Trimmed/*log .
$ multiqc .
Key Points
Adapter removal and trimming (optional) are important steps in processign RNA-seq data.