Quality control of the sequencing data.
Overview
Teaching: 30 min
Exercises: 0 minQuestions
What tools do we use to assess the data quality data in RNA-seq experiments?
Objectives
Learn how to assess data quality
Use the FastQC application to perform quality assessment
Use the MultiQC application to combine and view quality information
Several tools available to do quality assessemnt. For this workshop, we will use fastqc.
First, it is always good to verify where we are:
$ pwd
/home/[your_username]
# good I am ready to work
cd ~/obss_2021/
# copy the needed data into your directory
cp -r /nesi/projects/nesi02659/obss_2021/Resources/RNA_seq/ .
Checking to make sure we have the Raw files for the workshop.
$ ls
edna gbs genomic_dna intro_bash intro_r RNA_seq . ..
Creating a directory where to store the QC data:
$ cd RNA_seq
$ ls
Genome Raw rsmodules.sh yeast_counts_all_chr.txt
$ mkdir QC
Since we are working on the NeSI HPC, we need to search and load the package before we start using it.
Search
$ module spider fastqc
and then load
$ module purge
$ module load FastQC/0.11.9
hint : there is a file named rsmodules.sh which is a shell script to load the required modules at once. Running
source ~/RNA_seq/rsmodules.shcommand will excute it.
Now we can start the quality control:
$ fastqc -o QC/ Raw/*
You will see an automatically updating output message telling you the progress of the analysis. It will start like this:
Started analysis of SRR014335-chr1.fastq
Approx 5% complete for SRR014335-chr1.fastq
Approx 10% complete for SRR014335-chr1.fastq
Approx 15% complete for SRR014335-chr1.fastq
Approx 20% complete for SRR014335-chr1.fastq
Approx 25% complete for SRR014335-chr1.fastq
Approx 30% complete for SRR014335-chr1.fastq
Approx 35% complete for SRR014335-chr1.fastq
The FastQC program has created several new files within our RNA_seq/QC/ directory.
$ ls QC
SRR014335-chr1_fastqc.html SRR014336-chr1_fastqc.zip SRR014339-chr1_fastqc.html SRR014340-chr1_fastqc.zip
SRR014335-chr1_fastqc.zip SRR014337-chr1_fastqc.html SRR014339-chr1_fastqc.zip SRR014341-chr1_fastqc.html
SRR014336-chr1_fastqc.html SRR014337-chr1_fastqc.zip SRR014340-chr1_fastqc.html SRR014341-chr1_fastqc.zip
Viewing the FastQC results
If we were working on our local computers, we’d be able to look at each of these HTML files by opening them in a web browser.
However, these files are currently sitting on our remote NeSI HPC, where our local computer can’t see them. And, since we are only logging into NeSI via the command line - it doesn’t have any web browser setup to display these files either.
So the easiest way to look at these webpage summary reports will be to transfer them to our local computers (i.e. your laptop).
To transfer a file from a remote server to our own machines, we will use scp.
First we will make a new directory on our computer to store the HTML files we’re transferring. Let’s put it on our desktop for now. Open a new tab in your terminal program (you can use the pull down menu at the top of your screen or the Cmd+t keyboard shortcut) and type:
$ mkdir -p ~/Desktop/fastqc_html
$ scp -r [Your_UserName]p@login.mahuika.nesi.org.nz:~/obss_2021/RNA_seq/QC/ ~/Desktop/fastqc_html
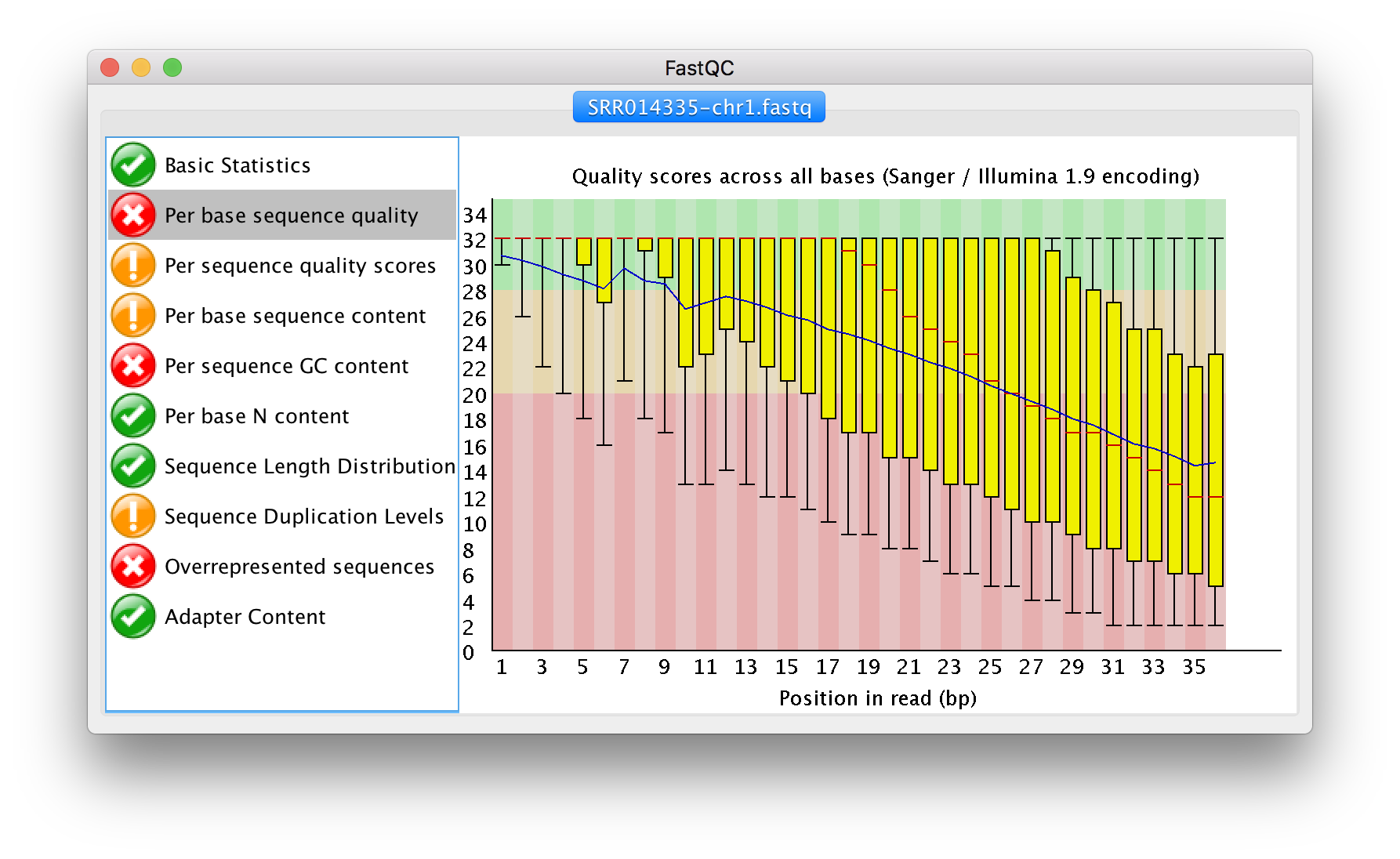
Working with the FastQC text output
Now that we’ve looked at our HTML reports to get a feel for the data, let’s look more closely at the other output files. Go back to the tab in your terminal program that is connected to NeSI and make sure you’re in our results subdirectory.
$ cd /home/fayfa80p/obss_2021/RNA_seq/QC
$ ls
SRR014335-chr1_fastqc.html SRR014336-chr1_fastqc.zip SRR014339-chr1_fastqc.html SRR014340-chr1_fastqc.zip
SRR014335-chr1_fastqc.zip SRR014337-chr1_fastqc.html SRR014339-chr1_fastqc.zip SRR014341-chr1_fastqc.html
SRR014336-chr1_fastqc.html SRR014337-chr1_fastqc.zip SRR014340-chr1_fastqc.html SRR014341-chr1_fastqc.zip
Let’s unzip the files to look at the FastQC text file outputs.
$ for filename in *.zip
> do
> unzip $filename
> done
Inside each unzipped folder, there is a summary text which shows results of the statistical tests done by FastQC
$ ls SRR014335-chr1_fastqc
fastqc_data.txt fastqc.fo fastqc_report.html Icons/ Images/ summary.txt
Use less to preview the summary.txt file
$ less SRR014335-chr1_fastqc/summary.txt
We can make a record of the results we obtained for all our samples by concatenating all of our summary.txt files into a single file using the cat command. We’ll call this fastqc_summaries.txt.
$ cat */summary.txt > ~/obss_2021/RNA_seq/QC/fastqc_summaries.txt
- Have a look at the fastqc_summaries.txt and search for any of the samples that have failed the QC statistical tests.
MultiQC - multi-sample analysis
- The FastQC analysis is applied to each sample separately, and produces a report for each.
- The application MultiQC provides a way to combine multiple sets of results (i.e., from MANY different software packages) across multipel samples.
- To generate
multqcresults, run the following command in the directory with the output files you want to summarise (e.g., fastqc reports generated above):
$ module load MultiQC/1.9-gimkl-2020a-Python-3.8.2
$ cd ~/obss_2021/RNA_seq/
$ mkdir MultiQC
$ cd MultiQC
$ cp ../QC/* ./
$ multiqc .
$ ls -F
multiqc_data/ multiqc_report.html
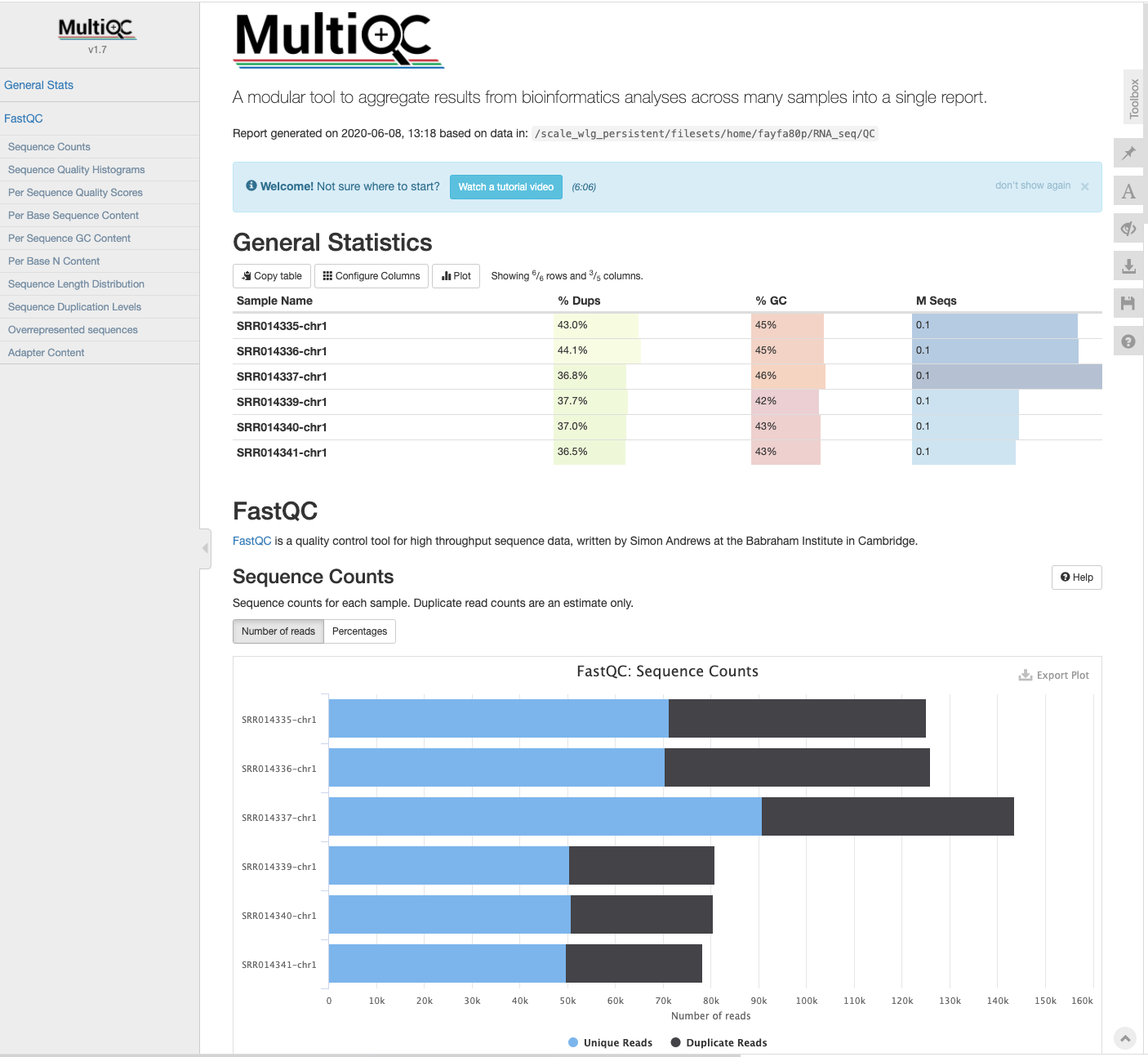
The html report shows the MultiQC summary
Key Points
Quality assessmet is a key initial step in the analysis of RNA-seq data
The FastQC and MultiQC applications are useful tools for quality assessent of RNA-seq data.