Introduction to Qiime
Overview
Teaching: 10 min
Exercises: 5 minQuestions
How do I import today’s results into Qiime?
Objectives
Identify the main components of metabarcoding and how to integrate them into a Qiime workflow
Overview
The Qiime2 package is an open-source system that incorporates multiple, stand-alone programs, giving you many options to run your analysis. The different programs are provided as plug-ins to provide maximum flexibility. The Qiime2 docs webpage has all the information you need to get started on processing metabarcoding data. There are multiple tutorials available, including a detailed overview of the available plugins and links to the concepts involved.
It is possible to run the entirety of your analysis within the Qiime2 system, but today we will just show how to import the results of the earlier lessons into the Qiime format, and then you will be able to follow most of the tutorials on the website.
First, here is a quick guided tour of the system.
Here is an overview of the Qiime2 workflow:
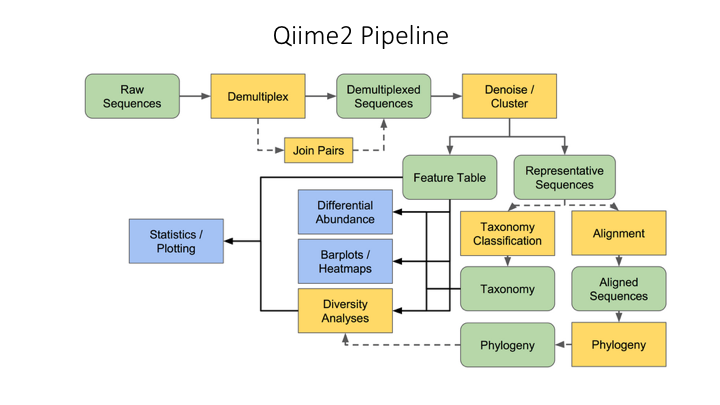
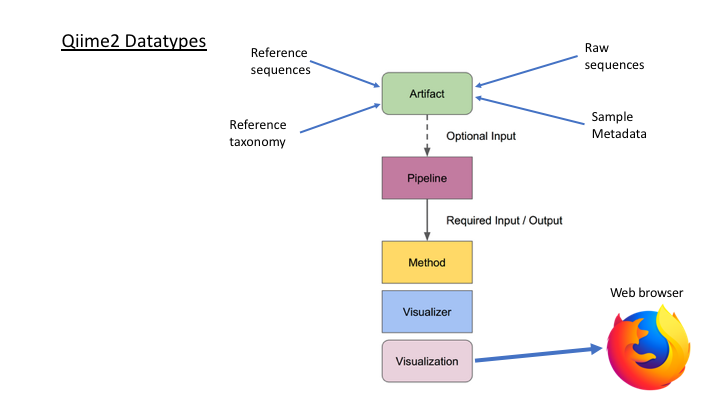
One of the main concepts of using Qiime is that all data is represented by what they refer to as “artifacts”, which are actually compressed folders that contain the data and metadata about the data. This is done so that all data files are tracked and logged through the system. In order to use Qiime data must be imported into their system; as well, to use the outputs of Qiime in other programs they must be exported (though some visuals provide a way to download flat files of some outputs).
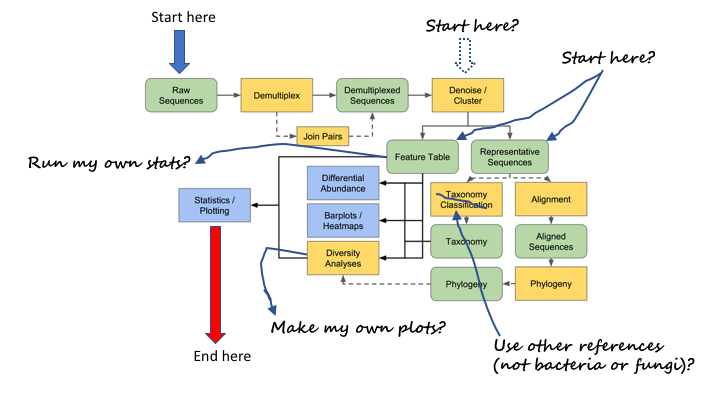
Often you do not want to start at the beginning or finish at the end of a pipeline. Once you are able to import today’s results, you will be able to start where you wish.
Tour of Qiime2 website
Here is a link to the resources on the Qiime2 website

Getting started: importing files into Qiime2 format
In the command line, we will first load the Qiime2 module:
$ module purge
$ module load QIIME2/2021.4
To see the options for any Qiime2 command, you can use help:
$ qiime tools import --help
There are just a few arguments for importing files, but many possible types and formats. Two of the commands are sort of help commands in that they show what is possible to import.
We can view those by just entering the option:
$ qiime tools import --show-importable-types
Sometimes it is necessary to specify the format of the input file. We can view those as well:
$ qiime tools import --show-importable-formats
These two arguments provide a good guide when you are trying to figure out the kind of file you are importing
Importing OTU fasta file and frequency table
To import the outputs of the Denoising and clustering lesson
module load QIIME2/2021.4
The Qiime import command (substitute the name of your otu file):
qiime tools import \
--type 'FeatureData[Sequence]' \
--input-path otus.fasta \
--output-path otus.qza
If the command/script worked then you should have a file called otus.qza in your /otus folder. All Qiime artifacts end in .qza, and all visuals end in .qzv.
We cannot import the frequency table directly into Qiime. First we have to convert our table into the biom format. Then the biom format will be imported to Qiime. Create a script called import_freq_table_to_qiime.sh and then put these commands:
biom convert -i otu_frequency_table.tsv \
-o fish_frequency.biom \
--to-hdf5
qiime tools import \
--input-path fish_frequency.biom \
--type 'FeatureTable[Frequency]' \
--input-format BIOMV210Format \
--output-path otu_frequency_table.qza
The output of this script should be two files: a .biom file and a qiime .qza frequency table file.
Importing taxonomy files into Qiime
import taxonomy output (with converter script)
Qiime has several methods to assign taxonomy to your OTUs (see Lesson 4). In order to run these, you must first convert the reference database to a Qiime flat file format, and then import it to Qiime. For the first part, you can use the tax_format tool in the Crabs program. If you have not done so, run the command to convert the reference file.
crabs_v1.0.1 tax_format \
--input ref_fasta.fasta \
--output ref_qiime \
--format qiif
You will notice that Crabs has split the original file into two separate files. One of these is a fasta file with the sequences and sequence ID, and the other is a table that has the taxonomic lineage for each sequence ID. Now you can import each of these files into Qiime.
qiime tools import \
--type 'FeatureData[Sequence]' \
--input-path qiime_ref.fasta \
--output-path qiime.refseqs.qza
qiime tools import \
--type 'FeatureData[Taxonomy]' \
--input-format HeaderlessTSVTaxonomyFormat \
--input-path qiime_ref.taxon \
--output-path qiime.taxon.qza
Now you are ready to assign taxonomy using either the BLAST or VSEARCH methods (see below). If you would like to run the Naive Bayes method, there is one further step, which is to train the sequences using a machine learning algorithm. However, as this takes time, one has been prepared for this course: lrRNA_fish_db_classifier.qza
For future reference, here is an example training command:
qiime feature-classifier fit-classifier-naive-bayes \
--i-reference-reads otus.qza \
--i-reference-taxonomy qiime.taxon.qza \
--o-classifier lrRNA_fish_db_classifier.qza \
--verbose
Using your imported files in Qiime
Now that you have all your outputs in Qiime format, you can use them for most of the tutorials on the Qiime web page.
Taxonomy assignment with Qiime
You can run three different methods in Qiime to do taxonomy assignment. For one example, see the basic taxonomic analysis tutorial. Below are examples for each of the methods, indicating which of our imported files to use.
BLAST Taxonomy Assignment
qiime feature-classifier classify-consensus-blast \
--i-query otus.qza \
--i-reference-reads qiime.refseqs.qza \
--i-reference-taxonomy qiime.taxon.qza \
--o-classification blast_output_taxonomy.qza \
--verbose
See the classify-consensus-blast plugin page for explanation of parameters and additional options
VSEARCH Taxonomy Assignment
qiime feature-classifier classify-consensus-vsearch \
--i-query otus.qza \
--i-reference-reads qiime.refseqs.qza \
--i-reference-taxonomy qiime.taxon.qza \
--o-classification vsearch_output_taxonomy.qza \
--verbose
See the classify-consensus-vsearch plugin page for explanation of parameters and additional options
Naive Bayes Taxonomy Assignment
qiime feature-classifier classify-sklearn \
--i-reads otus.qza \
--i-classifier lrRNA_fish_db_classifier.qza \
--o-classification nb_output_taxonomy.qza \
--verbose
See the classify-sklearn plugin page for explanation of parameters and additional options
Diversity analyses with Qiime
There are many analyses included in Qiime for doing community ecology statistics, including alpha and beta diversity. You can see an example in the Alpha and beta diversity analysis tutorial.
The main files that are used for most of Qiime’s diversity analyses are the frequency table and the sample metadata file. So, once you have imported the frequency table you can run many analyses (the sample metadata file is one of the few in the Qiime multiverse that remains in flat file format). Qiime has a pipeline command that will run multiple alpha and beta diversity metrics in one go, outputting everything to a separate folder.
The only other statistic that you will need for this is the sampling depth, which is the number that was used for rarefaction in Lesson 06 (90% of the lowest sample number, or XX)
qiime diversity core-metrics \
--i-table table.qza \
--p-sampling-depth XX \
--m-metadata-file sample_metadata.tsv \
--output-dir core-metrics-results
The outputs will all be in a folder called core-metrics-results.
Create a phylogeny from OTUs
There is one last example script. This is to align and create a phylogenetic tree from the OTUs. This can be done with separate commands, but Qiime provides a pipeline that 1) aligns the OTUs, 2) mask uninformative or ambiguous sites in the alignment, 3) infers a phylogenetic tree from the alignment, and then 4) roots the tree at the midpoint. You can check all the options at the Plugin page. You can also run these commands separately. There are other options for phylogeny, including inferring phylogeny with the iqtree and RaXMl programs. Have a look at all the options on the main Phylogeny plugin page. These tools can come in handy for multiple applications.
qiime phylogeny align-to-tree-mafft-fasttree \
--i-sequences otus.qza \
--o-alignment aligned-rep-seqs.qza \
--o-masked-alignment masked-aligned-rep-seqs.qza \
--o-tree unrooted-tree.qza \
--o-rooted-tree rooted-tree.qza
Once you have the phylogeny of the OTUs, you can run the diversity metrics again using unifrac distances:
qiime diversity core-metrics-phylogenetic \
--i-phylogeny rooted-tree.qza \
--i-table table.qza \
--p-sampling-depth XX \
--m-metadata-file sample-metadata.tsv \
--output-dir core-metrics-phylogenetic-results
Exporting your Qiime analysis out of Qiime
Qiime has many many tools that make it easy to analyse your data, but you may also want to use other programs, or analyse your outputs using Phyloseq or another statistical package in R or Python. Fortunately, Qiime provides a tool to export almost file you create in its format.
The Qiime export tool only allows you to export to a folder, so the best way is to first create a subfolder called exports:
mkdir exports
Then you will export whatever file you need to this subfolder:
qiime tools export \
--input-path INPUT-FILE \
--output-path exports
Qiime will export this file to the exports subfolder and give it a standard name. For example, if you export the results of a qiime taxonomy assignment, it will give it the name taxonomy.tsv. We advise changing this to a name specific to your analysis using bash mv:
mv exports/taxonomy.tsv qiime_tax_assign_table.tsv
Key Points
Once you import your results into Qiime, you can run any analysis with that program
The OTU fasta file that we created today are equivalent to ‘rep-seqs’ (representative sequences) in Qiime
The frequency table we created is called a ‘FeatureTable’ in Qiime parlance